
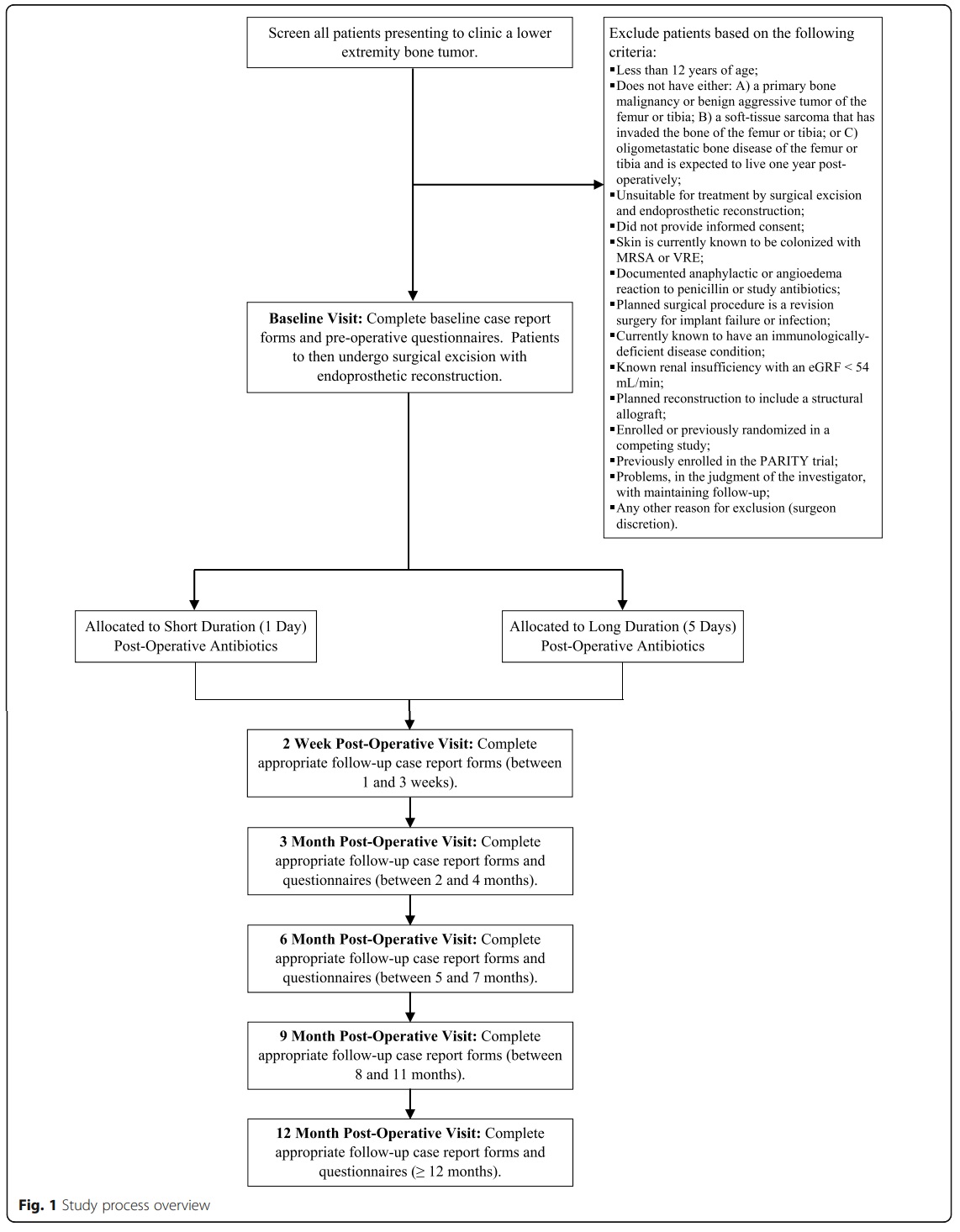
Background:
Limb salvage with endoprosthetic reconstruction is the current standard practice for the surgical management of lower extremity bone tumors in skeletally mature patients and typically includes tumor resection followed by the functional limb reconstruction with modular metallic and polyethylene endoprosthetic implants.
However, owing to the complexity and length of these procedures, as well as the immunocompromised nature of patients treated with chemotherapy, the risk of surgical site infection (SSI) is high. The primary research objective of the Prophylactic Antibiotic Regimens In Tumor Surgery (PARITY) trial is to assess whether a 5-day regimen of postoperative antibiotics decreases the risk of SSI at 1 year post-operatively compared to a 1-day regimen. This article describes the statistical analysis plan for the PARITY trial.
Methods/design:
The PARITY trial is an ongoing multi-center, blinded parallel two-arm randomized controlled trial (RCT) of 600 participants who have been diagnosed with a primary bone tumor, a soft tissue sarcoma that has invaded the bone or oligometastatic bone disease of the femur or tibia that requires surgical resection and endoprosthetic reconstruction. This article describes the overall analysis principles, including how participants will be included in each analysis, the presentation of results, adjustments for covariates, the primary and secondary outcomes, and their respective analyses. Additionally, we will present the planned sensitivity and sub-group
analyses.
Discussion:
Our prior work has demonstrated (1) high rates of SSI after the treatment of lower extremity tumors by surgical excision and endoprosthetic reconstruction, (2) highly varied opinion and practice among orthopedic oncologists with respect to prophylactic antibiotic regimens, (3) an absence of applicable RCT evidence, (4) extensive support from international investigators to participate in a RCT, and (5) the feasibility of conducting a definitive RCT to evaluate a 5-day regimen of post-operative antibiotics in comparison with a 1-day regimen.
Trial registration:
ClinicalTrials.gov NCT01479283. Registered on 24 November 2011
Keywords:
Orthopedic oncology, Bone sarcoma, Randomized controlled trial, Antibiotics, Statistical analysis plan
Background:
Limb salvage surgery is the current standard of care in the management of sarcoma of the long bones [1–3]. Advances in chemotherapeutic regimens and imaging
techniques allow for wide resection and functional reconstruction in 95% of patients. The most common type of long-bone reconstruction involves the use of a tumor
prosthesis or endoprosthesis. Due to the complexity and length of surgical resection and reconstruction, as well as the immunocompromised nature of patients treated
with chemotherapy, the risk of surgical site infection (SSI) remains high, which is a devastating complication that often requires staged revision surgery and longterm intravenous antibiotics [4, 5]. The risk for subsequent infection remains high, as does the risk for ultimate amputation [4, 5]. Moreover, patients’ quality-of-life and function following infection are dramatically impacted, as are healthcare costs [6, 7]. However, the most effective antibiotic regimen in preventing post-operative SSI remains controversial, and the current state of practice varies widely, particularly with respect to antibiotic duration [8]. Strategies to prevent SSIs and optimize quality-of-life while mitigating healthcare costs are needed.
The Prophylactic Antibiotic Regimens In Tumor Surgery (PARITY) trial is an ongoing international, multicenter randomized controlled trial (RCT) using a parallel two-arm design to determine whether a long duration (5 days) of post-operative prophylactic antibiotics decreases the risk of SSI when compared to a short duration (1 day) [9]. The protocol for the PARITY trial has been published elsewhere and provides more detail on the trial rationale, eligibility criteria, interventions, data management, and methods for minimizing bias [9]. Briefly, 600 participants 12 years of age or older undergoing surgical excision and endoprosthetic reconstruction of a lower extremity bone tumor across North America, South America, Europe, Australia, Africa, and Asia are randomized to receive either a short (1 day) or long (5 days) duration of post-operative antibiotics. Allocation is concealed using a centralized and automated 24-h computerized randomization platform that allows Internet-based randomization. Randomization is stratified by tumor location (femur or tibia) and clinical site in randomly permuted blocks of 2 and 4. The primary outcome of the study is the development of a SSI, guided by the Centers for Disease Control and Prevention (CDC) National Healthcare Safety Network reporting criteria [10]. Secondary outcomes include the development of antibiotic-related complications (such as gastrointestinal infections, fungal infections), unplanned re-operations, oncologic outcomes, mortality, and patient functional outcomes and quality-of-life at 1 year. Participants are regularly monitored post-operatively by the treating surgeon at 2 weeks, 6 weeks, 3 months, 6 months, 9 months, and 1 year following surgery. Outcome assessors and data analysts are blinded to treatment allocation. The full study
process is shown in Fig. 1.
In this article, we present our planned statistical analyses for the PARITY trial. The statistical analysis plan was finalized and approved on 11 January 2021 (version 1) for the PARITY trial protocol (31 October 2016, version 6) and in accordance with the trial master file, including the data management plan (11 December 2020, version 2). Ethics approval was granted for the Methods Centre at McMaster University (Hamilton Integrated Research Ethics Board No. 12-009) and at each participating clinical site (as per their local ethics committee). This trial is registered on ClinicalTrials.Gov (NCT01479283).
Methods
Outcomes
Primary outcome
The primary outcome of the PARITY trial is the development of a SSI within 1 year following the initial surgery to treat a lower extremity bone tumor. The primary
analysis is to assess whether a long duration regimen (5 days) of post-operative antibiotics decreases the risk of SSI at 1 year compared to a short duration regimen (1
day). SSIs are classified according to the criteria established by the CDC, which defines a SSI as an infection occurring within 30 days following the operative procedure or within 1 year if an implant is in place and the infection appears to be related to the procedure [10]. The SSI can involve any part of the body that is opened or manipulated during the operative procedure but excludes the skin incision, fascia, or muscle layers. The participant must also present with at least one of the following:
 Purulent drainage from the superficial/deep/organ space incision; Organisms isolated from an aseptically obtained culture of fluid or tissue from the superficial/deep/ organ space incision; Superficial/deep/organ space incision that is deliberately opened by a surgeon, attending physician, or other designee and is culture-positive OR not cultured and the participant has at least one of the following signs or symptoms: pain or tenderness, localized swelling, redness, or heat; or Diagnosis of a superficial/deep/organ space incisional SSI by a surgeon or attending physician.
Purulent drainage from the superficial/deep/organ space incision; Organisms isolated from an aseptically obtained culture of fluid or tissue from the superficial/deep/ organ space incision; Superficial/deep/organ space incision that is deliberately opened by a surgeon, attending physician, or other designee and is culture-positive OR not cultured and the participant has at least one of the following signs or symptoms: pain or tenderness, localized swelling, redness, or heat; or Diagnosis of a superficial/deep/organ space incisional SSI by a surgeon or attending physician.
Secondary outcomes
Secondary outcomes include the following:
1) Antibiotic-related complications including, but not limited to, Clostridioides difficile-associated colitis, opportunistic fungal infections, and indwelling catheter-related sepsis;
2) Unplanned re-operations including, but not limited to, amputation, irrigation and debridement, implant revision, and implant exchange;
3) Oncologic events;
4) All-cause mortality;
5) Physician-derived functional outcome as measured y the Musculoskeletal Tumor Society (MSTS)-87 and MSTS-93 scores; and
6) Self-reported functional outcome as measured by the Toronto Extremity Salvage Score (TESS) survey.
The MSTS-87 score is a standardized scoring system that is completed by an individual on the treatment team (preferably the treating surgeon) and measures physical
function after treatment for a musculoskeletal tumor across seven domains: motion, pain, stability, deformity, muscular strength, functional activity, and emotional acceptance. The MSTS-93 score is a standardized scoring system that is also completed by an individual on the treatment team and measures functional outcome after
treatment for a musculoskeletal tumor across six domains: pain, function, emotional acceptance, support, walking ability, and gait. The TESS survey is a selfadministered evaluation tool that was developed to assess physical function and quality-of-life in patients that have undergone limb salvage surgery for tumors of the extremities. The lower extremity portion of the survey contains 30 questions that are framed to ask about the difficulty experienced by the patient in performing each activity over the previous week. The MSTS-87, MSTS93, and TESS surveys are all commonly accepted functional scoring systems in the orthopedic oncology literature [11–13].
SSIs, antibiotic-related complications, re-operations, and mortality will be reviewed by an independent Adjudication Committee.
Sample size
At the onset of the trial, we calculated that the definitive trial’s sample size would include 460 participants per group, for a total of 920 participants. This sample size
was based on a between-group comparison for the primary outcome of deep SSI following long duration (5 days) or short duration (1 day) prophylactic antibiotics and was calculated to ensure that the study would have a power of 80% to identify differences among the two groups at an alpha level of 0.05, on the basis of an overall event rate of 10% and a presumed 50% or greater reduction in the risk of deep SSI within 1 year.
Prior to the transition from the pilot phase to the definitive phase of the trial, we met with our Steering Committee to finalize the definitive study protocol and processes. At this time, we decided to expand the trial’s primary outcome from deep SSI to any SSI (superficial/ deep/organ space SSI) in order to increase the expected event rate and study power without compromising clinical importance. This adjustment in the primary outcome resulted in an overall pilot phase event rate of 14%, which exceeds the overall event rate of 10% used to calculate the initial sample size. As a result, the definitive trial’s sample size was reduced to 300 participants per arm, for a total of 600 participants to identify the differences among the two groups at an alpha level of 0.05 and to ensure that the study would have a power of 80% using the updated event rate of 14% while maintaining the presumed 50% or greater reduction in the risk of SSI within 1 year.
The current sample size calculation is the standard method to determine sample size in a binary outcome study and will provide a conservative yet similar estimate to the more complicated and complex calculation for a time-to-event analysis. This decision was made as utilizing the more conservative binary outcome estimate would likely account for any unforeseen losses to followup, dropouts, and crossovers, which were considered negligible in our study population, and therefore, adjustments for their occurrences were not warranted at the time of the definitive sample size calculation. The binary outcome study method is also simpler to present in study documents such as grant applications.
Discussion
Analysis plan
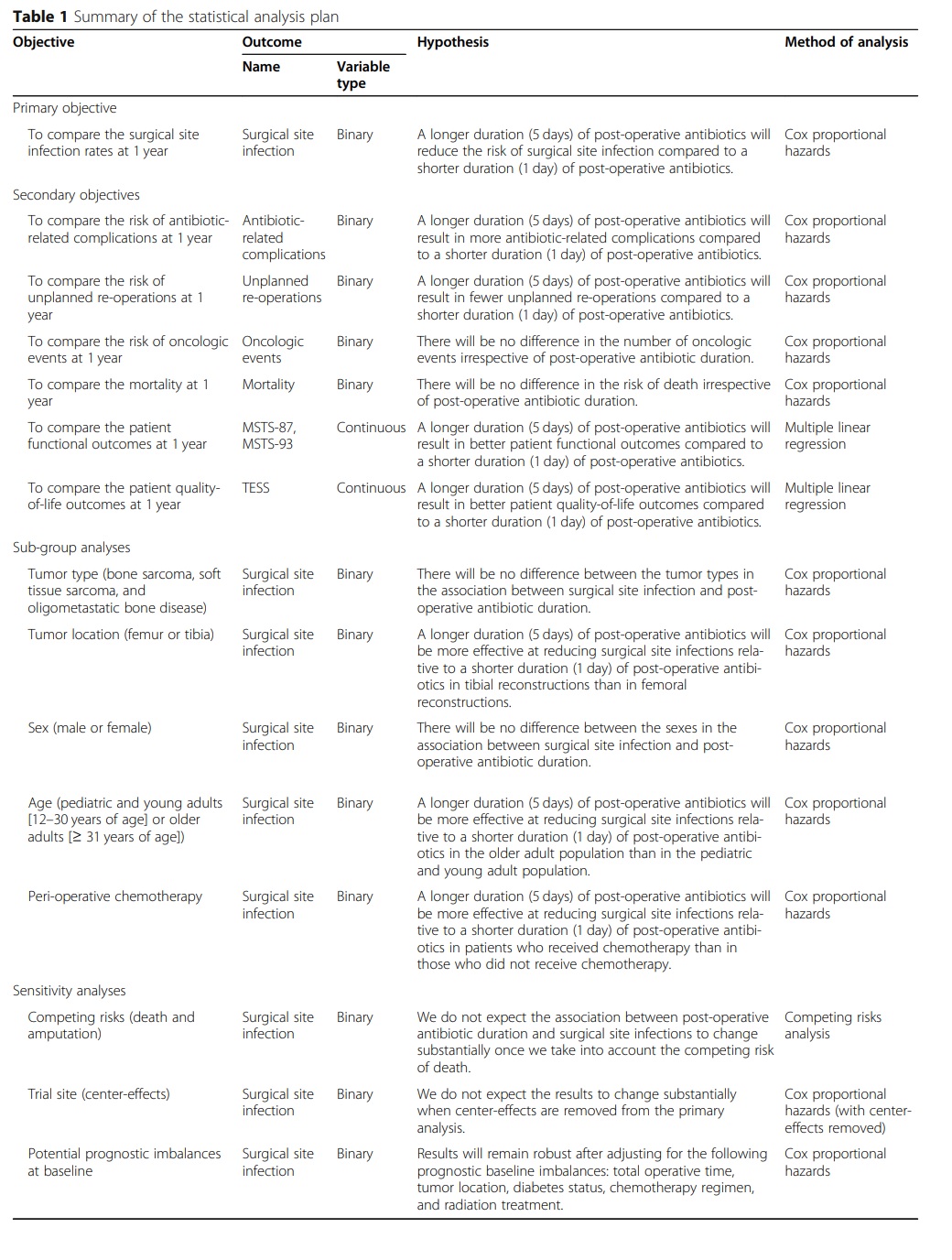
This statistical analysis plan follows the JAMA Guidelines for the Content of Statistical Analysis Plans in Clinical Trials [14]. A summary of all planned analyses is provided in Table 1.
Blinded analyses
All statistical analyses will first be conducted using blinded treatment groups (i.e., antibiotic duration X and duration Y). To do so, the blinded study statistician will
provide complete blinded results labeled antibiotic duration X and duration Y; the remainder of the study team at the Methods Centre will be left unaware of which
treatment groups antibiotic durations X and Y represent. Interpretations for the effect of the antibiotic durations will be documented during a blinded review of the data based upon blinded antibiotic duration X versus Y (i.e., we will determine how to interpret the results if antibiotic duration X proves to be the long duration regimen
(5 days) of post-operative antibiotics versus how to interpret the results if antibiotic duration Y proves to be the long duration regimen (5 days) of post-operative antibiotics) [15]. We will unblind the results by breaking the randomization code following the approval and documentation of the interpretations by the study team. These agreed-upon interpretations will guide the discussion section of the subsequent definitive trial manuscript.
Presentation of data
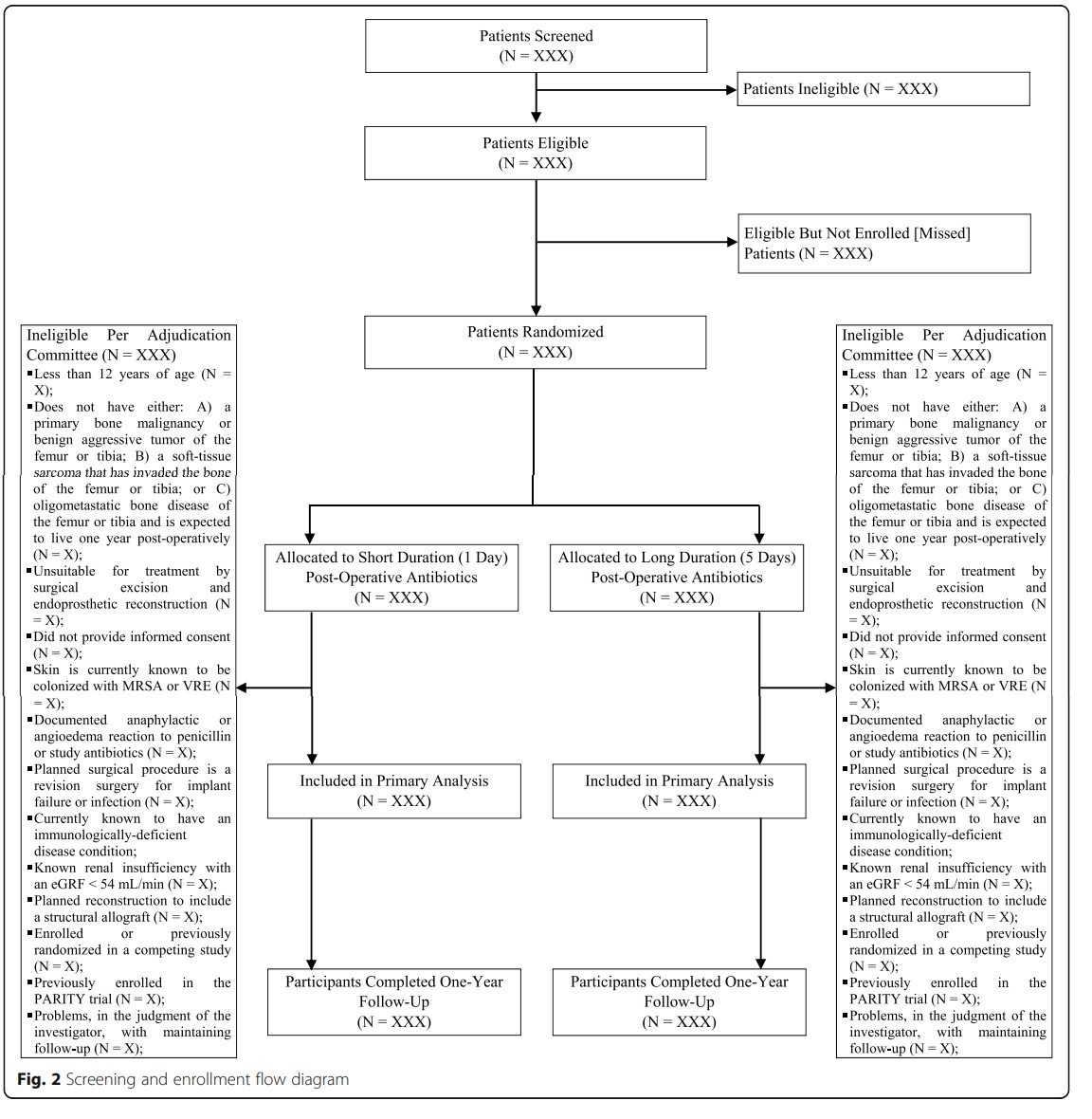
The trial results will be presented according to the Consolidated Standards of Reporting Trials (CONSORT) guidelines for RCTs [16]. The number of patients screened, included, and excluded will be presented in a flow diagram (Fig. 2). The baseline emographic characteristics, tumor details, and surgical and peri-operative management characteristics of the participants, as well as details of the prophylactic study antibiotic administrations, will be summarized by group. Continuous data will be presented with means and standard deviations [SD], or medians and first and third quartiles [Q1, Q3] for skewed data, and categorical data will be presented as frequencies and proportions (see Tables 2, 3, 4, and 5).
Primary outcome analysis
Our hypotheses for the primary analysis are as follows: Null hypothesis: There is no difference in the risk of SSI at 1 year between the two treatment groups. Alternate hypothesis: There is a difference in the risk of SSI at 1 year between the two treatment groups. The primary analysis will be a Cox proportional hazards analysis with time from surgery to the SSI as the primary outcome. Post-operative prophylactic antibiotic duration (treatment group [1 day versus 5 days]) will be the independent variable, and the Cox regression will also include tumor location (femur or tibia) and clinical site as stratification variables. All clinical sites with fewer than five participants enrolled will be collapsed into a single clinical site when included in our regression model. Participants who did not experience the primary endpoint will be censored at 12 months or the time of the last visit. The proportional hazards assumption of the Cox model will be assessed by examining Schoenfeld residuals. If the independent variable does not meet the assumption of the proportional hazards, we will modify the model to allow the HR to differ throughout the study period guided by the observed data. Results will be reported as HRs with the corresponding 95% CI and associated p values. Kaplan-Meier curves will be
constructed for the two randomized treatment groups. For each treatment group, we will also report superficial SSI, deep SSI, and organ space SSI. The results of the
primary analysis will be presented in Table 6.
Secondary outcomes analysis
We will estimate the effect of post-operative prophylactic antibiotic duration (1 day versus 5 days) on antibiotic-related complications, re-operations, oncologic events, and all-cause mortality at 1 year (Table 6).
Similar to the primary analysis, we will perform a Cox proportional hazards analysis. We will only perform Cox regressions for individual antibiotic-related complications, unplanned re-operations, and oncologic and mortality events if there are enough events. Should there be an insufficient number of events, we will summarize by treatment group and report using descriptive statistics (frequencies and proportions). We will also separately report 4-week, 3-month, and 1-year mortality figures for
future comparison with other studies.



In addition, we will also estimate the effect of postoperative prophylactic antibiotic duration on patient functional outcomes (MSTS-87 and MSTS-93 surveys) and quality-of-life (TESS surveys) at 1 year (Table 7). To do so, we will use multiple linear regression models that include the following independent variables: randomized treatment group, tumor location (femur versus tibia), clinical site, and baseline score. The results will be reported as the mean differences with 95% CIs. Given that functional and quality-of-life outcomes are the most difficult to collect and, therefore, we expect some missing data, we will use multiple imputation to address the missing data for these outcomes should the amount of missing data be considerable but not too substantial. Convention dictates that if more than five but less than 40% of data is missing, the use of multiple imputation is appropriate and warranted [17].
Sensitivity analyses
Sensitivity analyses will be performed for the primary outcome only [18]. We will conduct a competing risks analysis that accounts for deaths and amputation as
competing risks. We will also perform sensitivity analyses for center-effects where we will redo the primary analysis without including the clinical site in the model.
We will also look for prognostic imbalances between the two treatment groups based on the following key variables known to be risk factors for a SSI: total operative time, tumor location, diabetes status, chemotherapy regimen, and radiation treatment. We will complete adjusted analyses to address any possible baseline imbalance between the groups.

Sub-group analyses
At the onset of the PARITY trial, we identified two important sub-groups (tumor type and tumor location), which will be reported according to the standard guidelines [19]. As we near the end of the trial, prior to unblinding, we have identified a further three important sub-groups (sex, age, and peri-operative chemotherapy). We will add a main effect for the sub-group variable and the treatment by sub-group interaction to our primary model described above to assess whether the magnitude of the treatment effect is significantly different between the sub-groups (Fig. 3). This will be repeated separately for each sub-group variable. We will perform the following sub-group analyses with the primary endpoint as the outcome (Fig. 3):
1) Tumor type—the type of tumor will be classified as follows: bone sarcoma, soft tissue sarcoma, or oligometastatic bone disease. We hypothesize that there will be no difference between the tumor types with regard to the association between prophylactic antibiotic duration and risk of infection.
2) Tumor location—the location of the tumor will be classified as follows: femur or tibia (we will not include the stratification variable of tumor location in this analysis). We hypothesize that a long duration (5 days) of prophylactic antibiotics will be more effective relative to a short duration (1 day) in tibial reconstructions than in femoral
reconstructions.

3) Sex—sex will be classified as follows: male or female. We hypothesize that there will be no difference between the sexes with regard to the association between prophylactic antibiotic duration and risk of infection.
4) Age—age will be classified as follows: pediatric and young adults (12–30 years of age) or older adults (≥ 31 years of age). We hypothesize that a long duration (5 days) of prophylactic antibiotics will be more effective relative to a short duration (1 day) in the older adult population than in the pediatric and young adult population.
5) Peri-operative chemotherapy—peri-operative chemotherapy will be classified as follows: no chemotherapy or chemotherapy (neoadjuvant or adjuvant or a combination of the two). We hypothesize that a long duration (5 days) of prophylactic antibiotics will be more effective relative to a short duration (1 day) in patients who received chemotherapy than in those who did not receive chemotherapy.
Rather than pre-specifying a threshold p value for making a sub-group claim, we will use the approach suggested by Sun et al. to consider the plausibility of any possible subgroup effects [20]. If a plausible sub-group effect is found, we will further explore the impact of the sub-group on the secondary outcomes. However, due to inadequate sample size and power to conduct the sub-group analyses, these results will be used solely for the generation of hypotheses for further investigations.
Interim analyses
No interim analyses are planned due to our desire to avoid spuriously inflated estimates of treatment effects [21, 22]. The PARITY Data and Safety Monitoring Board (DSMB) regularly meets to monitor the study data for participant safety.
Dissemination
Upon trial completion, the primary manuscript with the 1-year follow-up results, whether positive, negative, or neutral, will be submitted for peer review and publication in a top-tier medical journal. The final dataset will be shared through an open access data repository once all analyses are completed.
Trial status
The PARITY trial began as a pilot of 60 participants in January 2013 [23]. Upon demonstrating study feasibility and securing definitive funding (July 2014), these participants were rolled into the definitive study (N = 600). Recruitment for the definitive study was completed in October 2019, and the final 1-year follow-up data is expected to be completed and collected in December 2020.
Abbreviations
CDC: Centers for Disease Control and Prevention; CI: Confidence interval;
CONSORT: Consolidated Standards of Reporting Trials; DSMB: Data and Safety
Monitoring Board; HR: Hazards ratio; ITT: Intention-to-treat;
MSTS: Musculoskeletal Tumor Society; PARITY: Prophylactic Antibiotic
Regimens In Tumor Surgery; RCT: Randomized controlled trial; TESS: Toronto
Extremity Salvage Score; SD: Standard deviation; SSI: Surgical site infection
Acknowledgements
Full authorship list for the PARITY Investigators:
Steering Committee: Michelle Ghert (Chair, McMaster University), Mohit Bhandari (Co-Chair, McMaster University), Benjamin Deheshi (McMaster University), Gordon Guyatt (McMaster University), Ginger Holt (Vanderbilt University Medical Center), Timothy O’Shea (McMaster University), R. Lor Randall (University of California at Davis Medical Center), Lehana Thabane (McMaster University), Roberto Vélez (Hospital Vall d’Hebron), and Jay Wunder (Mount Sinai Hospital).
Methods Centre: Michelle Ghert [principal investigator]; Patricia Schneider, Victoria Giglio, and Paula McKay [project management]; Sheila Sprague [research methodologist]; Diane Heels-Ansdell [statistical analysis]; and Lisa Buckingham [data management] (McMaster University).
Data and Safety Monitoring Board: Peter Rose (Chair, The Mayo Clinic), Brian Brigman (Duke University Medical Center), and Eleanor Pullenayegum (The Hospital for Sick Children).
Adjudication Committee: Michelle Ghert (Chair, McMaster University), Timothy O’Shea (McMaster University), R. Lor Randall (University of California at Davis Medical Center), and Robert Turcotte (McGill University Health entre).
Participating Clinical Sites:
Juravinski Hospital and Cancer Centre (Hamilton, ON, Canada)—Michelle Ghert (PI), Benjamin Deheshi, and David Wilson
Mount Sinai Hospital (Toronto, ON, Canada)—Peter Ferguson (PI) and Jay Wunder
McGill University Health Centre (Montreal, QC, Canada)—Robert Turcotte (PI) and Krista Goulding
The Ottawa Hospital (Ottawa, ON, Canada)—Joel Werier (PI) and Hesham Abdelbary Vancouver General Hospital (Vancouver, BC, Canada)—Paul Clarkson Hôpital Maisonneuve-Rosemont (Montreal, QC, Canada)—Marc Isler (PI) and Sophie Mottard
CHUQ – L’Hôtel-Dieu de Québec (Québec City, QC, Canada)—Norbert Dion (PI) and Annie Arteau
Vanderbilt University Medical Center (Nashville, TN, USA)—Ginger Holt (PI), Jennifer Halpern, and Herbert Schwartz
Beth Israel Deaconess Medical Center (Boston, MA, USA)—Megan Anderson (PI) and Mark Gebhardt
Boston Children’s Hospital (Boston, MA, USA)—Megan Anderson (PI) and Mark Gebhardt
Huntsman Cancer Institute (Salt Lake City, UT, USA)—Kevin Jones (PI) and R. Lor Randall
Memorial Sloan-Kettering Cancer Center (New York, NY, USA)—John Healey (PI) Hospital Universitario Austral (Buenos Aires, ARG)—Marcos Galli Serra (PI) Royal Adelaide Hospital (Adelaide, SA, AUS)—Mark Clayer (PI)
University of Connecticut Health Center (Farmington, CT, USA)—Adam Lindsay (PI) and Tessa Balach
The Rothman Institute (Philadelphia, PA, USA)—John Abraham (PI) and Scot Brown
Holden Comprehensive Cancer Center (Iowa City, IA, USA)—Benjamin Miller (PI) University of Minnesota Medical Center (Minneapolis, MN, USA)—Edward Cheng (PI)
Wexner Medical Center (Columbus, OH, USA)—Thomas Scharschmidt (PI) and Joel Mayerson
Emory University Orthopedics and Spine Center (Atlanta, GA, USA) – Nickolas Reimer (PI)
Montefiore Medical Center (New York, NY, USA)—David Geller (PI) and Bang Hoang
Stanford University Hospital and Clinics (Palo Alto, CA, USA)—Raffi Avedian (PI) Foothills Medical Centre (Calgary, AB, Canada)—Shannon Puloski (PI) and Michael Monument
Sinai Hospital of Baltimore (Baltimore, MD, USA)—Albert Aboulafia (PI) SUNY Upstate University Hospital (East Syracuse, NY, USA)—Timothy Damron (PI)
Maimonides Medical Center (New York, NY, USA)—Howard Goodman (PI)University of Pittsburgh Medical Center (Pittsburgh, PA, USA)—Kurt Weiss (PI) and Mark Goodman
Massachusetts General Hospital (Boston, MA, USA)—Joseph Schwab (PI)
Franklin Square Medical Center (Baltimore, MD, USA)—Albert Aboulafia (PI)
Albany Medical Center (Albany, NY, USA)—Matthew DiCaprio (PI) and Bradford Palmer
Oregon Health and Science University Hospital (Portland, OR, USA)—Yee-Cheen Duong (PI), Kenneth Gundle, and James Hayden
Johns Hopkins Hospital (Baltimore, MD, USA)—Carol Morris (PI) and Adam Levin
Grey’s Hospital (Pietermaritzburg, ZAF) – Reitze Rodseth (PI) and Leonard Marais Instituto de Ortopedia e Traumatologia da Universidade de São Paulo (São Paulo, BRA)—André Mathias Baptista (PI) and Juan Pablo Zummaraga
Hospital Vall d’Hebron (Barcelona, ESP)—Roberto Vélez (PI) University of California at San Francisco Medical Center (San Francisco, CA, USA)—Rosanna Wustrack (PI), and Richard O’Donnell
Cincinnati Children’s Hospital (Cincinnati, OH, USA)—Joel Sorger (PI) University of Maryland Medical Center (Baltimore, MD, USA)—Daniel Lerman (PI)
University of Florida Health Shands Hospital (Gainesville, FL, USA)—André Spiguel (PI), C. Parker Gibbs, and Mark Scarborough
Leiden University Medical Center (Leiden, NLD)—P.D. Sander Dijkstra (PI) and Michiel van de Sande
All India Institute of Medical Sciences (New Delhi, IND)—Shah Alam Khan (PI) and Venkatesan Sampath Kumar
Medical College of Wisconsin (Milwaukee, WI, USA)—John Neilson (PI) Long Island Jewish Medical Center [Northwell Health] (New Hyde Park, NY, USA)—Howard Goodman (PI)
Dartmouth-Hitchcock Medical Center (Hanover, NH, USA)—Eric Henderson (PI)
Saint Louis University Hospital (St. Louis, MO, USA)—David Greenberg (PI)
University Medical Center Groningen (Groningen, NLD)—Paul Jutte (PI)
The Cleveland Clinic (Cleveland, OH, USA)—Nathan Mesko (PI) and Lukas Nystrom
Children’s Cancer Hospital Egypt (Cairo, EGY)—Ahmed Elghoneimy (PI) Hartford Hospital (Hartford, CT, USA)—Adam Lindsay (PI)
Hospital de Clínicas de Porto Alegre (Porto Alegre, BRA)—Ricardo Becker (PI)
University of Arkansas for Medical Sciences (Little Rock, AR, USA)—Richard Nicholas (PI)
University of California at Los Angeles Medical Center (Los Angeles, CA, USA)—Nicholas Bernthal (PI), Jeffrey Eckhardt, and Francis Hornicek
Medical University Graz (Graz, AUT)—Andreas Leithner (PI) and Marko Bergovec
Singapore General Hospital (Singapore, SNG)—Mann Hong Tan (PI) and Suraya Zainul Abidin
University of California at Davis Medical Center (Sacramento, CA, USA)—Steven Thorpe (PI) and R. Lor Randall
Authors’ contributions
Together, PS and DHA drafted the initial statistical analysis plan. PS drafted the manuscript and incorporated all author edits. DHA critically revised the manuscript for important intellectual content. LT provided important intellectual content to the initial statistical analysis plan and critically revised the manuscript for important intellectual content. MG made substantial contributions to the conception and design of the statistical analysis plan, critically revised the manuscript for important intellectual content, and has greed to be accountable for all aspects of the work. The PARITY Investigators contributed to the design, conduct, and overall data collection for the PARITY trial. All authors have read and approved the final manuscript to be published.
Funding
Research grants to conduct this research were received from the following funding sources: Canadian Cancer Society Research Institute (CCSRI) Innovation and Innovation to Impact Grants (PI: M. Ghert), Canadian Institutes of Health Research (CIHR) Operating Grant (PI: M. Ghert, M. Bhandari), Canadian Orthopedic Foundation (COF) J. Édouard Samson Award, Orthopedic Research and Education Foundation/Musculoskeletal Tumor Society (OREF/MSTS) Clinical Research Grant (PI: M. Ghert), and Physicians’ Services Incorporated (PSI) Health Research Grant (PI: M. Ghert). The funding sources had no role in the design or conduct of the study; the collection, management, analysis, or interpretation of the data; or the preparation, review, or approval of the manuscript. Dr. M. Ghert had full access to all of the study data and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Availability of data and materials
The final dataset will be shared through an open access data repository once all analyses are completed.
Declarations
Ethics approval and consent to participate
Ethics approval, including Informed Consent Form approval, was granted for the Methods Centre at McMaster University by the Hamilton Integrated Research Ethics Board (HiREB No. 12-009), as well as at each participating clinical site as per their local ethics committee.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Author details
1 Department of Surgery, McMaster University, Hamilton, ON L8V 1C3, Canada. 2 Department of Health Research Methods, Evidence and Impact, McMaster University, Hamilton L8S 4L8, ON, Canada. 3 Juravinski Hospital and Cancer Centre, Hamilton Health Sciences, 711 Concession Street, B3 169A, Hamilton L8V 1C3, ON, Canada.